Introducción
¿Sabías que algunas dermatitis en manos pueden no deberse a productos químicos, sino a proteínas naturales como frutas o látex?
Presentamos un caso clínico de dermatitis de contacto proteica (DCP) en el contexto laboral, una entidad poco frecuente pero relevante en la práctica clínica de dermatólogos y alergólogos. El caso ilustra la importancia de una anamnesis detallada, la exploración dirigida y el papel clave de la colaboración interdisciplinar en el diagnóstico y manejo de estas dermatosis ocupacionales.Caso clínico
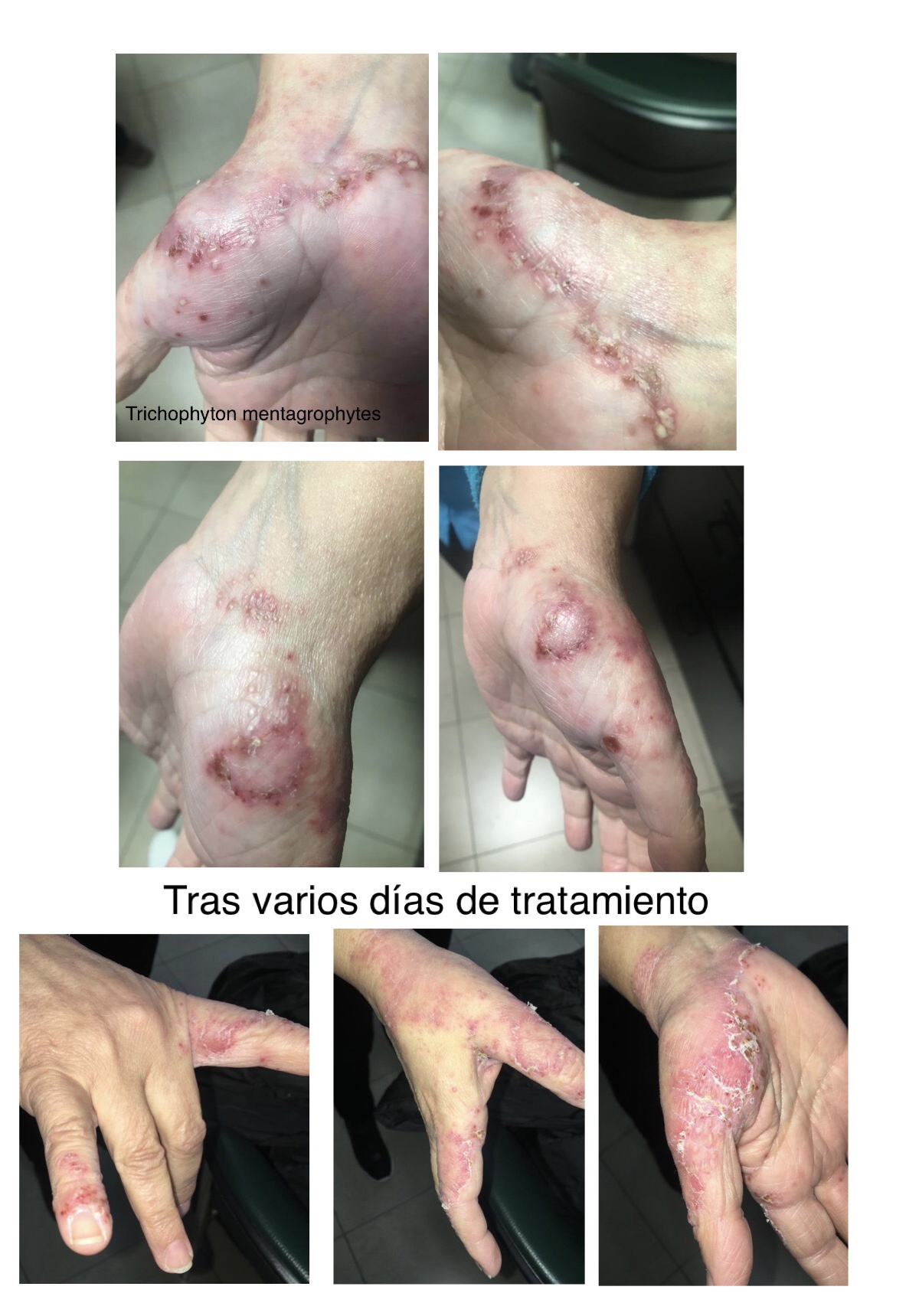
Mujer de 47 años, remitida a alergología por eccema crónico. Con antecedentes de dermatitis alérgica de contacto por sensibilización a múltiples alérgenos (níquel, thiuram, carbas, etilendiamina). Presenta eccema crónico en manos, antebrazos, escote y cara. Empeoramiento de síntomas vinculado a actividades laborales con exposición a frutas y verduras.
Historia laboral diversa con exposición en cocinas y frutería. Utiliza doble guante de protección. Actualmente de baja médica. Exploración revela lesiones eccematosas con signos de cronicidad.


Exploraciones complementarias y diagnóstico
Pruebas epicutáneas positivas para mezclas de thiuram, carba y níquel. Pruebas prick positivas a alimentos y neumoalérgenos. Determinación ISAC e IgE total elevada.
Diagnóstico: dermatitis de contacto proteica ocupacional con sensibilización combinada inmediata y retardada. Se establece relación clara con entorno laboral.
prueba como herramienta clave en la evaluación de sospechas clínicas de DCP.
Fisiopatología y epidemiología
La DCP combina mecanismos inmunológicos tipo I y tipo IV. Las proteínas de alto peso molecular presentes en alimentos o sustancias naturales actúan como alérgenos. Afecta principalmente a trabajadores en contacto directo con productos frescos, látex o enzimas. En España, las enfermedades dermatológicas representan una proporción significativa de enfermedades profesionales.
La DCP combina mecanismos inmunológicos tipo I y tipo IV. Las proteínas de alto peso molecular presentes en alimentos o sustancias naturales actúan como alérgenos. Afecta principalmente a trabajadores en contacto directo con productos frescos, látex o enzimas. En España, las enfermedades dermatológicas representan una proporción significativa de enfermedades profesionales.
Diagnóstico diferencial y herramientas diagnósticas
El diagnóstico se basa en la historia clínica, exploración física y pruebas cutáneas específicas (prick, parche). La evaluación laboral y la asociación temporal entre exposición y síntomas son claves.
El diagnóstico se basa en la historia clínica, exploración física y pruebas cutáneas específicas (prick, parche). La evaluación laboral y la asociación temporal entre exposición y síntomas son claves.
Estrategias terapéuticas
Incluyen la evitación del alérgeno, tratamiento tópico (corticoides, inhibidores de calcineurina, emolientes), tratamiento sistémico (antihistamínicos, corticoides, inmunomoduladores), educación del paciente y seguimiento clínico. Además, se enfatiza la necesidad de evaluación de riesgos laborales y uso de equipos de protección adecuados.
Manejo por parte de dermatología en el manejo de DCP ocupacional
La dermatitis de contacto proteica (DCP) requiere un enfoque multidisciplinar, donde la colaboración entre dermatología y alergología resulta esencial. Mientras que el dermatólogo lidera la evaluación clínica cutánea, el diagnóstico diferencial con otras dermatosis, y el tratamiento tópico y preventivo, el alergólogo complementa el estudio mediante pruebas de hipersensibilidad inmediata (prick-test, IgE específica) para identificar reacciones tipo I. Esta coordinación permite un abordaje integral del paciente, optimizando tanto el diagnóstico como el manejo terapéutico y legal, especialmente en contextos de exposición laboral donde se requiere documentación precisa para el reconocimiento como enfermedad profesional.
Avances en fisiopatología e investigación
Nuevas evidencias implican células NK, dendríticas y citocinas, ampliando dianas terapéuticas. Interés en microbioma cutáneo, genética, epigenética e inmunoterapia. Se identifican líneas futuras de estudio para un abordaje más preciso de la dermatitis alérgica.
Legislación relevante
La legislación española reconoce la DCP como enfermedad profesional si se demuestra su relación directa con la actividad laboral. Normativa aplicable: Ley 31/1995, RD 39/1997, RD 486/1997, RD 374/2001 y RD 1299/2006 (Código 2D0101).
Conclusiones
DCP debe sospecharse ante eccema refractario en trabajadores expuestos a productos frescos.
El diagnóstico exige pruebas tanto de hipersensibilidad inmediata como retardada.
La coordinación con salud laboral es crucial para el reconocimiento como enfermedad profesional
El diagnóstico exige pruebas tanto de hipersensibilidad inmediata como retardada.
La coordinación con salud laboral es crucial para el reconocimiento como enfermedad profesional
1. Johansen JD, AaltoKorte K, Agner T, Andersen KE, Bircher A, Bruze M, et al. European Society of Contact Dermatitis guideline for diagnostic patch testing—recommendations on best practice. Contact Dermatitis. 2015;73(4):195–221.
2. Dickel H, Mahler V, Nast A, Bauer A, Becker D, Brasch J, et al. German S1 guideline: Contact dermatitis. J Dtsch Dermatol Ges. 2022;20(1):1–20.
3. Rustemeyer T, et al. Mechanisms of irritant and allergic contact dermatitis. In: Contact Dermatitis, 5th ed. Cham: Springer; 2021. p. 43–90.
4. Schal H, et al. Protein contact dermatitis: Review of 27 cases. Actas DermosiSifiliográficas (English Edition). 2011;102(7):587–594.
By Remedios Taberner Hermosilla, MIR de Medicina del Trabajo en D.S. Castelló. Julio 2025









%20.JPG)